【遺伝子導入(DNAトランスフェクション)】
培養細胞へ目的の遺伝子を導入する方法としては、リン酸Ca法、Lipofection法、エレクトロポレーション法等があります。ここでは手法が比較的容易であり、一般に広く用いられているLipofection法について解説します。
 原理
原理
生体膜を構成しているリン脂質は、水溶液中ではliposomeと呼ばれる脂質2重膜の小胞を形成します。Lipofection法は、この小胞を利用して培養細胞内へcDNAを導入する方法で、cDNAをliposomeと混合することにより、liposome小胞内にcDNAを封入して細胞と培養すると、liposome膜が細胞膜へ融合しcDNAが細胞質内に入り、核内に移動することによって、cDNAの発現が起こると考えられています。
我々の研究室ではQIAGEN社のPolyFect Transfection Reagentを使用しています。
培養細胞
一般的にはHEK、COS、CHO細胞などが用いられています。
我々の研究室ではtsA201という細胞を用いています。tsA201細胞はHEK293細胞由来の株で、細胞の形状がスペースクランプに適しているため、速いボルテージクランプが必要な電位依存性ナトリウムチャネル等の解析に従来用いられています。
プラスミド
我々の研究室ではQIAGEN社のDNA精製キット(midi、maxiprep)により精製したプラスミドを用いています。また発現ベクターは、pIRES2-EGFP(CLONTECH社)またはpcDNA3(Invitrogen社)を用いています。
DNAトランスフェクションプロトコル
概ねマニュアル通りですが、我々の研究室での一例です。
1)トランスフェクションの前日あるいは前々日に35mmのディッシュに細胞をまきます。
2)細胞を37℃、5%CO2でインキュベートします。トランスフェクションの当日に培養ディッシュの50~80%程度が細胞で覆われている状態が理想的です。
3)エッペンチューブに血清、タンパク質および抗生物質を含まない培養液100μlを入れ、そこへ滅菌水に溶かしたDNA2μgを加え希釈します。
4)PolyFect Transfection Reagent20μlを(3)のDNA溶液に添加します。ピペットで5、6回アップダウンを行いミックスします。
5)PolyFect ReagentとDNA溶液の複合体形成のために室温で5~10分間インキュベートします。
6)複合体形成中に(2)の培養ディッシュの培養液を新しい培養液(血清と抗生物質を含む)1.5ml交換します。
7)細胞培養液(血清と抗生物質を含む)0.6mlを(5)のトランスフェクション複合体を含むチューブに添加します。ピペットで2、3回アップダウンしてミックス後、すぐにトランスフェクション混合液をすべて(6)の35mmの培養ディッシュ中の細胞へ添加します。ディッシュを静かに回して、複合体を均質に行き渡らせます。
8)(7)のトランスフェクション複合体と細胞を37℃、5%CO2でインキュベートし、遺伝子発現を促します。この場合使用している細胞系および導入した遺伝子にもよるのですが、我々の研究室では6時間程度のインキュベーションを行っています。
9)適切な時間のインキュベーションを行った後、(8)のディッシュから液を静かに吸引除去し、2mlのPBS(-)で一度ウォッシュします。そこへ0.01%Trypsin-EDTA(tsA201の場合)0.2mlを加え、37℃、5%CO2中に約1分間置いて細胞を剥がします。
10)新しい35mmのディッシュ3枚を用意して、そこへ細胞培養液(血清と抗生物質を含む)を2ml入れておく。
11)(9)に細胞培養液(血清と抗生物質を含む)3mlを加えてよく混ぜたら、(10)のディッシュに1mlずつ入れて行き、細胞が均質になるようにディッシュを振り(タテタテヨコヨコがおすすめ)ます。
この割合は細胞の状態などによって、適宜変更してみて下さい。
12)発現させる遺伝子、細胞の状態などで様々ですが、一般的には翌日から電気生理実験が可能となります。
【電気生理学的測定】
ここではパッチクランプアンプとしてEPC9、ソフトウェアはPulse(ともにHEKA社)を使用して、電圧固定法によるWhole-cell記録を例に、哺乳類培養細胞を用いたパッチクランプ測定法について解説します。


 1)最初にEPC9アンプボックスの電源を入れてから、コンピュータを起動します。
1)最初にEPC9アンプボックスの電源を入れてから、コンピュータを起動します。
 2)次にPULSEプログラムを立ち上げます。プログラムを立ち上げると、PULSEを選ぶかPULSEFIT(データ解析用ソフト)を選ぶかの選択画面が現れるので、PULSEを選択する。もし、電源が入っていなかったり、EPC9が接続されていなかったりすると警告が出るのでやり直します。
2)次にPULSEプログラムを立ち上げます。プログラムを立ち上げると、PULSEを選ぶかPULSEFIT(データ解析用ソフト)を選ぶかの選択画面が現れるので、PULSEを選択する。もし、電源が入っていなかったり、EPC9が接続されていなかったりすると警告が出るのでやり直します。
 3)初期化に成功すると、新しいデータファイルを作る"Create"か前のファイルにデータを追加していく"Modify"かを聞かれます。通常、実験の始めには日付を入れた新しいデータファイルを"Create"しておくと良いと思います。
3)初期化に成功すると、新しいデータファイルを作る"Create"か前のファイルにデータを追加していく"Modify"かを聞かれます。通常、実験の始めには日付を入れた新しいデータファイルを"Create"しておくと良いと思います。
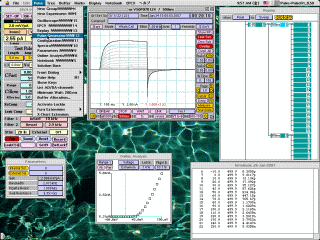 4)立ち上げると、デフォルトではEPC9アンプの制御画面とオシロスコープウィンドウが現れます。他にも表示させたいウィンドウがあればメニューの"Pulse"から必要なものを選ぶことができます。
4)立ち上げると、デフォルトではEPC9アンプの制御画面とオシロスコープウィンドウが現れます。他にも表示させたいウィンドウがあればメニューの"Pulse"から必要なものを選ぶことができます。
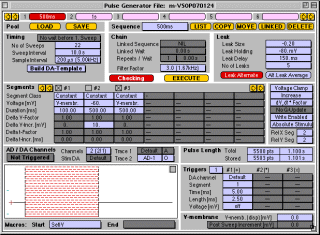 5)メニューの"Pulse"からPulse Generator画面を表示させて、取り込み条件の設定、パルスプロトコルを作成します。作成したプロトコルは図のそれぞれのボタンに登録することができます。よく使用するプロトコルはこの中に登録しておくと便利です。ここではHolding
Potential-60mVから500msで10mVのStep Pulseを+150mVまで10S間隔で記録するプロトコルを設定しています。(Leak
SubtractionはP/n法を用いて、目的とするパルスの後に小さい逆向きのパルスをいくつか重ね合わせることによって、余分な成分、リーク電流を差し引くものです。)
5)メニューの"Pulse"からPulse Generator画面を表示させて、取り込み条件の設定、パルスプロトコルを作成します。作成したプロトコルは図のそれぞれのボタンに登録することができます。よく使用するプロトコルはこの中に登録しておくと便利です。ここではHolding
Potential-60mVから500msで10mVのStep Pulseを+150mVまで10S間隔で記録するプロトコルを設定しています。(Leak
SubtractionはP/n法を用いて、目的とするパルスの後に小さい逆向きのパルスをいくつか重ね合わせることによって、余分な成分、リーク電流を差し引くものです。)
 6)電極内液の充填
6)電極内液の充填
記録する細胞を探したら、細胞内液を充填したガラス電極を電極ホルダーに装着します。初めに電極の先端を内液の入ったチューブに浸し、毛細管現象を利用して先端側から内液を電極にほんの少し入れます。(この操作により、電極の先端にゴミがつまるのを防ぐことができます。)続いて、細い注射針(28ゲージ)または先を細長く伸ばしたピペットチップを用いて反対側から内液を注入します。その際、気泡が入ってしまったら、指ではじいて追い出しておきます。(小さな泡が電極の先に残っていないかよく確認します。)
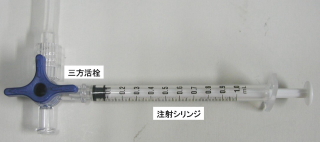 7)電極を細胞外液に浸ける
7)電極を細胞外液に浸ける
内液を充填した電極を電極ホルダーに装着したら、細胞外液中に浸す前に軽く陽圧をかけておきます。(ディスポの注射シリンジ1mlと三方活栓を組み合わせた吸引システムが良いと思います。この場合、0.2~0.3ml程度。)
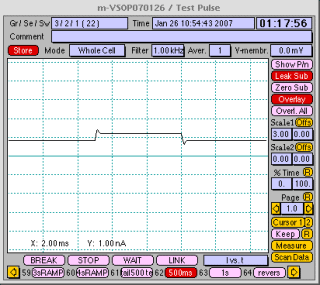 陽圧をかけながら電極を外液中に入れると、図のようにオシロスコープウィンドウに矩形波が表示されます。
陽圧をかけながら電極を外液中に入れると、図のようにオシロスコープウィンドウに矩形波が表示されます。
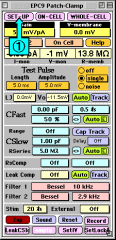 ここで①の"Setup"ボタンを押すと、アンプのゲイン設定と"Test Pulse"の大きさが初期化されて、
ここで①の"Setup"ボタンを押すと、アンプのゲイン設定と"Test Pulse"の大きさが初期化されて、
 液間電位などのオフセットが自動補正されます。
液間電位などのオフセットが自動補正されます。
8)ギガオームシールを作る
マニピュレータの粗動操作により電極を細胞に近づけます。先端が細胞の真上にくるようにします。次にマニピュレータの微動操作により、電極をさらに細胞に接近させます。電極が細胞にあたり、そこに少し”くぼみ”ができたところでストップします。電極が細胞に触れたかどうかは電極を流れる電流量が変化するので、抵抗値の変化からも知ることができます。次に陽圧を解除すると、細胞が電極に張り付き、この操作だけでギガシールが形成されることもありますが、大抵はその後に引き続き、徐々に少しずつ陰圧をかけて細胞を吸引していくことでギガオームシールが完成されます。ギガシール形成後は過度に電極内に細胞膜を吸い込むことを防ぐため、陰圧は開放しておきます。
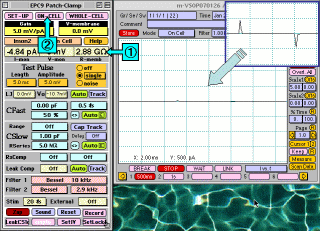 9)ホールセル記録をする
9)ホールセル記録をする
シールの抵抗値は、コントロールパネル①のR-membraneに表示されている値から確認します。この状態で②の"On Cell"ボタンを押すと、On
Cellモードになり、ガラス電極の持つ浮遊容量(C-Fast)が補正されます。
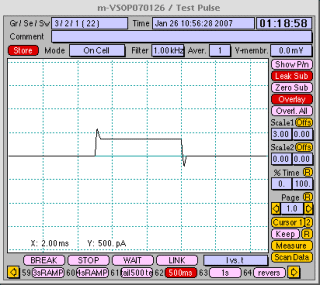
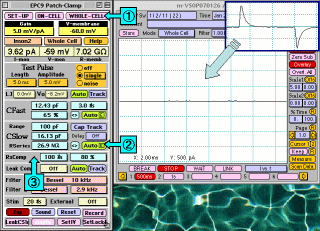 ここからさらにゆっくりと陰圧(吸引)をかけていくと、パッチ膜が破れて再び一過性の容量性電流が観察されます。(右上の囲み図)ここで①の"WHOLE
CELL"ボタンを押してWHOLE CELLモードにします。次にコントロールパネル②のCSlow"Auto"ボタンを押し、細胞膜容量を補正します。あわせて、③のRsCompによりシリーズレジスタンスの補正を行います。(通常は時定数100μsで補正率70~90%ぐらい。発振しない程度に。)
ここからさらにゆっくりと陰圧(吸引)をかけていくと、パッチ膜が破れて再び一過性の容量性電流が観察されます。(右上の囲み図)ここで①の"WHOLE
CELL"ボタンを押してWHOLE CELLモードにします。次にコントロールパネル②のCSlow"Auto"ボタンを押し、細胞膜容量を補正します。あわせて、③のRsCompによりシリーズレジスタンスの補正を行います。(通常は時定数100μsで補正率70~90%ぐらい。発振しない程度に。)
後はいよいよ実際に作成したパルスプロトコルを使って、記録を行うだけです。さあ始めてみましょう!
Copyrightc2009 NIPS Technical Division. All Rights Reserved.