2細胞器官研究系
2.1生体膜研究部門
生体膜研究部門は2007年6月に新たな研究体制(深田正紀教授)となった。本研究部門では脳の情報処理メカニズムを分子レベルで解明し、また、その破綻がどのようにして‘てんかん’や‘アルツハイマー病’等の疾患をもたらすのかを明らかにする。
AMPA型グルタミン酸受容体は神経活動に応答してダイナミックにシナプスに輸送され、脳内の主要な興奮性神経伝達を司る。このAMPA受容体のシナプスでの発現量の変化がシナプスに可塑的変化をもたらす。したがって、AMPA受容体がどのようにしてシナプス膜へ輸送され、その機能が制御されているかは現在の神経科学における重要な命題である。当研究部門では最近独自に同定した1)パルミトイル化脂質修飾酵素P-PAT、および2)てんかん関連新規リガンドLGI1を起点としてAMPA受容体を介したシナプス伝達の制御機構を統合的に解明することを目指している。
1.パルミトイル化脂質修飾酵素P-PATによるAMPA受容体動態制御機構の解明
私たちはAMPA受容体をシナプスにアンカリングする足場蛋白質PSD-95に焦点を当て研究をおこなっている。最近、私たちはPSD-95のシナプス膜への局在を制御するパルミトイル化脂質酵素(P-PAT)を同定し、P-PATがPSD-95のパルミトイル化を介してAMPA受容体のシナプス膜での発現量を調節することを明らかにした。2007年はP-PAT酵素の活性が神経活動により制御されるかどうかを検討し、その活性制御機構について研究をおこなった。海馬培養神経細胞を用いてパルミトイル化PSD-95を測定、可視化する方法を開発し、パルミトイル化修飾を受けたPSD-95の変動およびAMPA受容体の動態を検討した。P-PATの活性は神経活動により負に調節され、AMPA受容体の恒常性維持に関わっていることを明らかにした。すなわち、「神経活動の抑制→P-PATの活性化→PSD-95のパルミトイル化レベルの上昇→ポストシナプスPSD-95量増加→AMPA受容体のシナプスへの集積→神経活動の維持」というフィードバック機構が存在することを明らかにした。
2.てんかん関連新規リガンドLGI1によるAMPA受容体制御機構の解明
私たちは最近、PSD-95に結合する蛋白質をラット脳から精製し、LGI1、ADAM22およびStargazinを同定した。これら3つの蛋白質は遺伝学的にいずれもてんかんに関連した蛋白質であった。また、分泌蛋白質LGI1はADAM22と結合することによりAMPA受容体の機能を促進することを明らかにした。2007年はLGI1の作用機構を明らかにする目的でLGI1複合体を脳内から精製し、MudPIT(Multidimensional Protein Identification Technology)法にて高感度、高い特異性でもってLGI1複合体構成蛋白質を複数同定した。さらに、LGI1の個体レベルでの機能を明らかにする目的でLGI1のノックアウトマウス作成に着手し、現在キメラマウスを得ている。このように、生化学的な側面と遺伝学的な側面から新規リガンドLGI1によるAMPA受容体制御機構とてんかん発症メカニズムにアプローチする基盤が整いつつある。
図 AMPA受容体動態制御モデル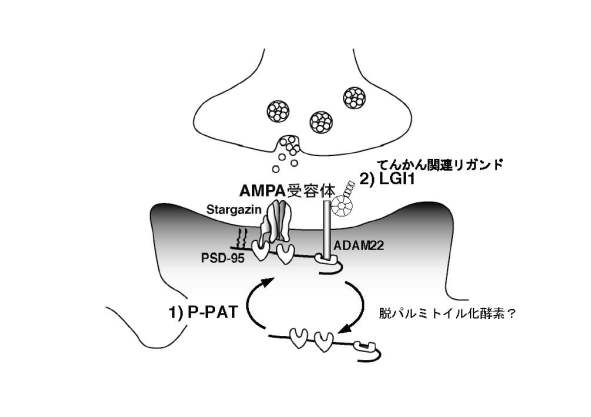
(1)パルミトイル化酵素P-PATはPSD-95のパルミトイル化を介してPSD-95のシナプス後部膜(ポストシナプス)局在を規定する。PSD-95は足場蛋白質としてstargazinを介してAMPA型グルタミン酸受容体の足場となり、AMPA受容体のシナプス後部膜での発現量を制御している。最近、P-PATの活性は神経活動により制御され、AMPA受容体の恒常性維持に重要な役割を担っていることを明らかにした。(2)一方、新規リガンド・受容体LGI1/ADAM22は、PSD-95と結合してAMPA受容体機能を制御するが、その分子機構は解析中である。
2.2機能協関研究部門
私達の部門は1992年以来、容積調節や吸収・分泌機能や環境情報受容などのようにすべての細胞種が持っている最も一般的で基本的な細胞機能とそのメカニズムを、チャネル、トランスポータ、レセプターなどの膜機能分子の働きとして統合的に解明すると共に、細胞死誘導のメカニズムをそれらの異常として把握することを目標に研究している。2007年度は、主として次の3研究課題に取組んだ。
1. 細胞容積調節の分子メカニズムと高浸透圧誘導性カチオンチャネル
細胞はその容積を正常に維持する能力を持ち、膨張後の調節はRegulatory Volume Decrease(RVD)、縮小後の調節はRegulatory Volume Increase(RVI)と呼ばれる。これらのメカニズムには種々のチャネルやトランスポータやレセプターの働きが関与する。RVIをもたらすNaCl流入路は、高浸透圧刺激によって活性化されるカチオンチャネルHypertonicity-Induced Cation Channel (HICC)、Na+/H+アンチポータ(NHE)とCl-/HCO3-アンチポータ(AE)によって与えられることがわかっている。本年度はこのHICCの活性化に関与するシグナルメカニズムを調べ、チロシンキナーゼ、p38-MAPキナーゼ、蛋白質キナーゼCやホスフォリパーゼCなどの関与を明らかにした(Wehner et al, 2007 Cell Physil Biochem)。また、このHICCの新しいブロッカー(2-APB)を発見した(Numata et al, 2007 J Physiol Sci)。
2. 細胞死の誘導メカニズムと細胞容積調節破綻
アポトーシスの初期過程に伴われる持続性の容積縮小はApoptotic Volume Decrease(AVD)、ネクローシスに伴われる持続性膨張はNecrotic Volume Increase(NVI)と呼ばれ、これらのメカニズムにも種々のチャネルやトランスポータが関与する。ミトコンドリア仲介性アポトーシス刺激の場合も、デスレセプター仲介性アポトーシス刺激の場合も、抗癌剤シスプラチンによる癌細胞のアポトーシスの場合にもAVDを実現するための細胞外Cl-流出を担うアニオンチャネルは容積感受性外向整流性Cl-チャネル(VSOR)であることをこれまで明らかにしてきた。本年度は、一過性前脳虚血によって海馬CA1ニューロンにもたらされる遅発性神経細胞死にもVSORが関与し、そのブロッカーのin vivo投与によってこれが救済されることを明らかにした(Inoue et al, 2007 J Neurosci Res)。更には、アポトーシス死時に生ずる細胞内ATP濃度の増加が解糖系反応の亢進によることを証明した(Zamaraeva et al, 2007 Biochem Biophys Res Commun)。そして、これまでの私達の研究結果を総括して、持続的細胞縮小化そのものがアポトーシス誘導に本質的かつ不可欠の条件を与えることを明確にした(Shimizu et al, 2007 Acta Physiol Sinica)。 ニューロンのグルタミン酸過興奮毒性によるネクローシス死にもNVIが伴われることが知られている。このときに発生する細胞体膨張や樹状突起varicosity形成によって活性化されるVSORは、グルタミン酸除去後にはRVDをもたらして細胞死救済的に働くが、グルタミン酸刺激持続下ではグルタミン酸レセプター活性化によってもたらされる脱分極によってCl-流入の経路を与えることになって細胞死誘導的に働くことをはじめて明らかにした(Inoue & Okada, 2007 J Neurosci)。
3. ストレスシグナル伝達と細胞外シグナル放出性アニオンチャネル
ストレス時に細胞から放出されたATPが細胞間シグナル伝達に重要な役割を果たすことは、多くの組織で知られている。その放出機序としてはエキソサイトーシス性のものとそうでないものがあるが、後者へのマキシアニオンチャネルの関与をこれまでに証明してきた。本年度は、グリア細胞においても、マキシアニオンチャネルが虚血性ATP放出にも(Liu et al, 2007 Purinergic Signal)、低浸透圧性ATP放出にも(Liu et al, 2007 Cell Res)関与することを明らかにした。心筋細胞のマキシアニオンチャネルの分布は、ATP放出サイトの分布とよく一致すること、そして成熟ラット心筋細胞の場合にはT管開口部に限局することをスマートパッチ法を用いて明らかにした(Dutta et al, 2007 Biophys J)。