
Abstract
We will clarify the molcecular mechanism on synapic development and plasticity by focusing on 1) dynamic protein palmitoylation and 2) synaptic protein complex.
Molecular mechanism for dynamic protein palmitoylation
Posttranslational processing events, including phosphorylation, ubiquitinylation and lipid modification, provide a central molecular mechanism for cells to respond to extracellular signals.
Protein palmitoylation -the frequent lipid modification with the lipid palmitate- regulates protein trafficking and function.
Palmitoylation modifies numerous classes of proteins including synaptic vesicle proteins, synaptic scaffolding proteins, signaling proteins and cytoskeletal proteins to target them to the specialized microdomain of the plasma membrane.
Palmitoylation is unique in that it is reversible and dynamically regulated by specific extracellular signals.
However, the molecular mechanism of protein palmitoylation has been unknown due to the difficulty in enzymology of protein palmitoylation and visualization of palmitoylated state of proteins in cells.
To overcome these difficult but important problems, we will reveal the regulatory mechanism of recently isolated enzyme family that mediates palmitoylation.
We have recently isolated the novel Palmitoyl-Acyl Transferase family (Fukata M et al. Neuron 44: 987-996), and identified PSD-95 palmitoylating enzymes (named P-PAT).
Importantly, this novel enzyme family has substrate specificity.
We also found that inhibition of P-PAT activity in neurons reduced palmitoylation and synaptic clustering of PSD-95 and diminished AMPA receptor-mediated neurotransmission.
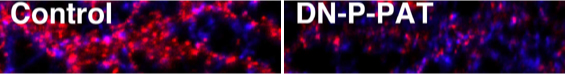
|
Red: Surface AMPA receptors
Blue: Synaptophysin |
Synaptic protein complexes
Dynamic regulation of AMPA receptors (AMPARs) underlies aspects of synaptic plasticity.
Although numerous AMPAR-interacting proteins have been identified, their quantitative and relative contributions to native AMPAR complexes remain unclear.
We quantitated protein interactions with neuronal AMPARs by immunoprecipitation from brain extracts.
We found that stargazin-like transmembrane AMPAR regulatory proteins (TARPs) co-purified with neuronal AMPARs, but we found negligible binding to GRIP1, PICK1, NSF, or SAP-97.
To facilitate purification of neuronal AMPAR complexes, we generated a transgenic mouse expressing an epitope-tagged GluR2 subunit of AMPARs.
Taking advantage of this powerful new tool, we isolated two populations of GluR2 containing AMPARs: an immature complex with the ER chaperone BiP, and a mature complex containing GluR1, TARPs and PSD-95. (Fukata Y et al. J. Cell Biol. 2005)
Recently, we purified PSD-95-associated synaptic protein complex from rat brain and identified a complex comprising stargazin, LGI1 and ADAM22.
Importantly, all these major components are genetically linked to epilepsy.
Stargazin is mutated in stargazer mice with absence epilepsy and ataxia.
LGI1 is a secreted neuronal protein and its mutations have been found in patients with familial epilepsy.
ADAM22 is a transmembrane protein and ADAM22-deficient mice were reported to lead to death from seizures.
We found that LGI1 is an extracelluar ligand for ADAM22, whereas the mutated form of LGI1 in patients with familial epilepsy fails to bind to ADAM22.
ADAM22 links extracellular LGI1 to PSD-95-scaffolding platform containing AMPAR and stargazin.
Furthermore, we found LGI1 and ADAM22 interaction specifically augments AMPAR-mediated excitatory synaptic transmission in hippocampal slices.
This study identifies a novel pathway in which LGI1, an extracellularly-released neuronal protein, controls synaptic strength and indicates possible avenues for understanding human epilepsy (Fukata, Y. et al., Science 313:1792-1795, 2005).
We are now investigating (i) if and how neuronal LGI1 secretion is regulated by synaptic activity; (ii) the signaling events downstream of LGI1/ADAM22 interaction; and (iii) the physiological and pathological roles of LGI1/ADAM22 in synaptic transmission and epileptogenesis.