Cardiac plasticity is defined as structural remodeling of the heart in response to environmental demands and is an intrinsic compensatory mechanism to reduce hemodynamic workload. Exercise, pregnancy, and postnatal growth all promote physiologic myocardial growth, whereas neurohumoral activation, hypertension, and myocardial injury can all cause pathological hypertrophy, which increases the risk of heart failure by reducing myocardial flexibility through myocardial stiffening. Cardiac atrophy, defined as an acquired reduction in the size and mass of the heart, is a complication of protracted bed rest, prolonged weightlessness during space travel, and mechanical unloading through the use of a ventricular assist device. The atrophied myocardium also exhibits reduced contractility due to decreased myocardial flexibility. Why myocardial flexibility is reduced during the processes of both cardiac atrophy and pathological hypertrophy is largely unknown.
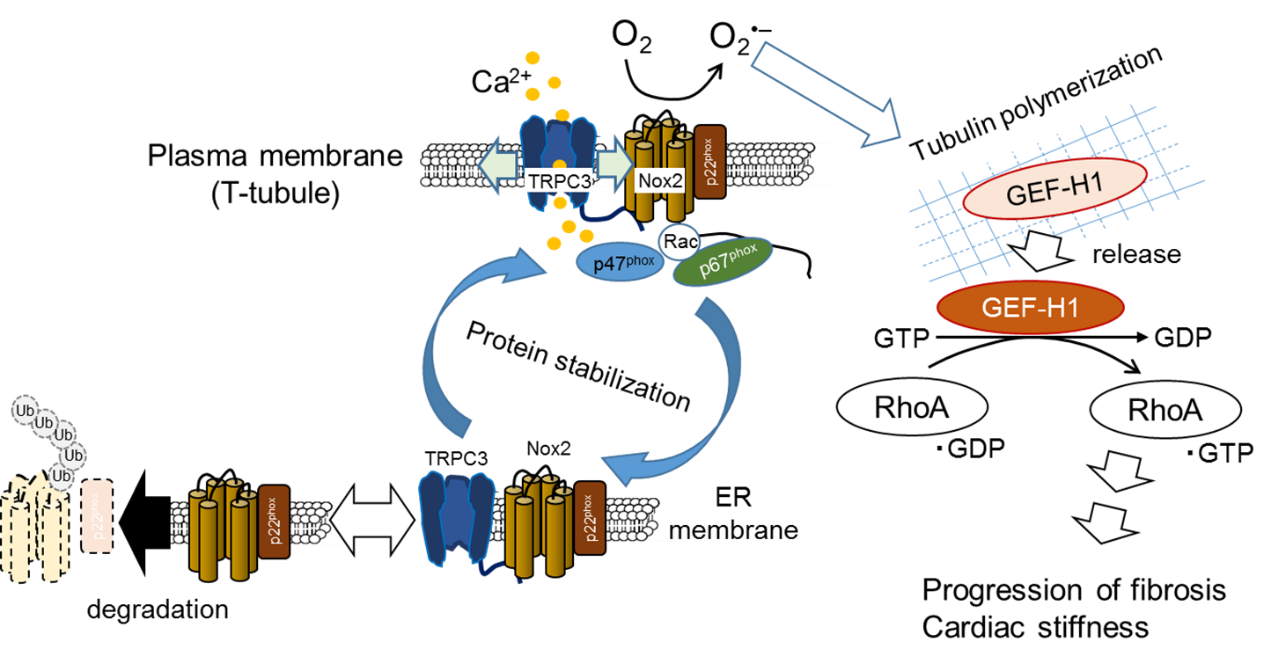
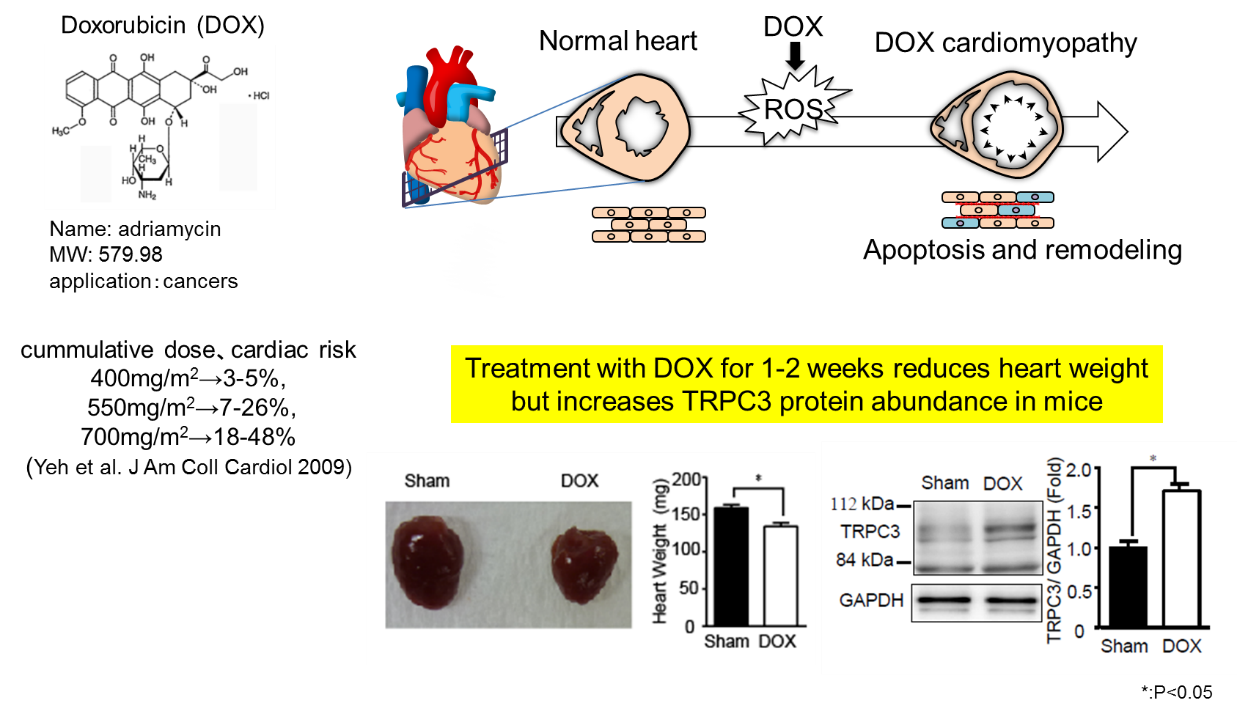
Doxorubicin (DOX) is a highly effective anticancer agent used to treat a variety of hematologic and solid malignancies. However, its dose-dependent cardiotoxicity limits its clinical use. DOX initially forces the heart to shrink, which leads to induction of myocardial apoptosis and interstitial fibrosis at later stages of LV dilated cardiomyopathy. There is growing evidence that the DOX-induced cardiac apoptosis and fibrosis are mainly caused by excess production of reactive oxygen species (ROS), which is initiated by NADPH oxidase (Nox) activation. We have previously reported that transient receptor potential canonical 3 (TRPC3) channels forms protein complex with Nox2 to stabilize and increase ROS generating activity in cardiac myocytes and fibroblasts, which leads to induction of interstitial fibrosis (i.e., deposition of collagen type I and III) and myocardial stiffness in mouse left ventricular (LV) myocardium (Figure 1).
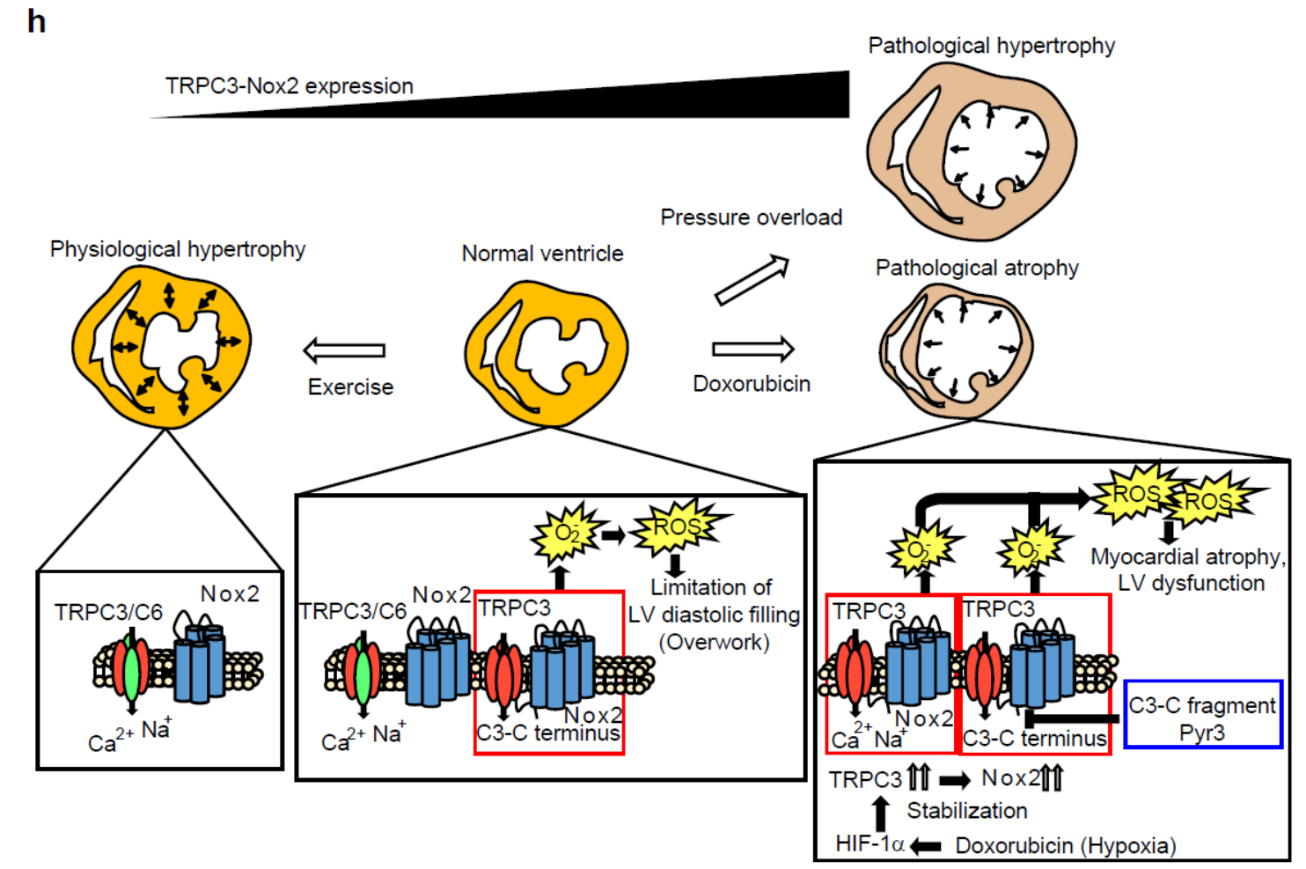
Here, we demonstrate that inhibiting transient receptor potential canonical 3 (TRPC3) channels abolishes doxorubicin-induced myocardial atrophy in mice. Doxorubicin increased production of reactive oxygen species (ROS) in rodent cardiomyocytes through hypoxic stress-mediated upregulation of NADPH oxidase 2 (Nox2), which formed a stable complex with TRPC3. Cardiomyocyte-specific expression of TRPC3 C-terminal mini-peptide inhibited TRPC3-Nox2 coupling and suppressed doxorubicin-induced reduction of myocardial cell size and LV dysfunction and its upregulation of Nox2 and oxidative stress, without reducing hypoxic stress (Figure 2).
Voluntary exercise, an effective treatment to prevent doxorubicin-induced cardiotoxicity, also downregulated the TRPC3-Nox2 complex and promoted volume load-induced LV compliance, as demonstrated in TRPC3-deficient hearts (Figure 3). These results illustrate the impact of TRPC3 on LV compliance and flexibility and, focusing on the TRPC3-Nox2 complex, provide a new strategy for prevention of doxorubicin-induced cardiomyopathy.
This research was funded by grants from the Japan Science and Technology Agency, Precursory Research for Embryonic Science and Technology Program (JPMJPR1336 to M.N.); Grants-in-Aid for Scientific Research (No. 25293018 and 16776376 to M.N.) and Scientific Research on Innovative Areas (Research in a Proposed Research Area ‘Oxygen Biology’ (No. 14430291 to M.N.) and ‘Living in Space’ (No. 16H01656 to T.N-T.) and Platform for Drug Discovery, Informatics, and Structural Life Science (to M.N.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N, Uchida K, Sumimoto H, Mori Y and Nishida M.
TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy.
JCI Insight. 2017;2(15):e93358. https://doi.org/10.1172/jci.insight.93358.
[Ref.]
Kitajima N. et al., TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 6, 37001; doi: 10.1038/srep37001 (2016).
Numaga-Tomita, T. et al., TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 6, 39383; doi: 10.1038/srep39383 (2016).
![]()
![]()
![]()
![]()
![]()
National Institute for Physiological Sciences
Gunma University
Kyushu University
Kyoto University
Tokyo University





