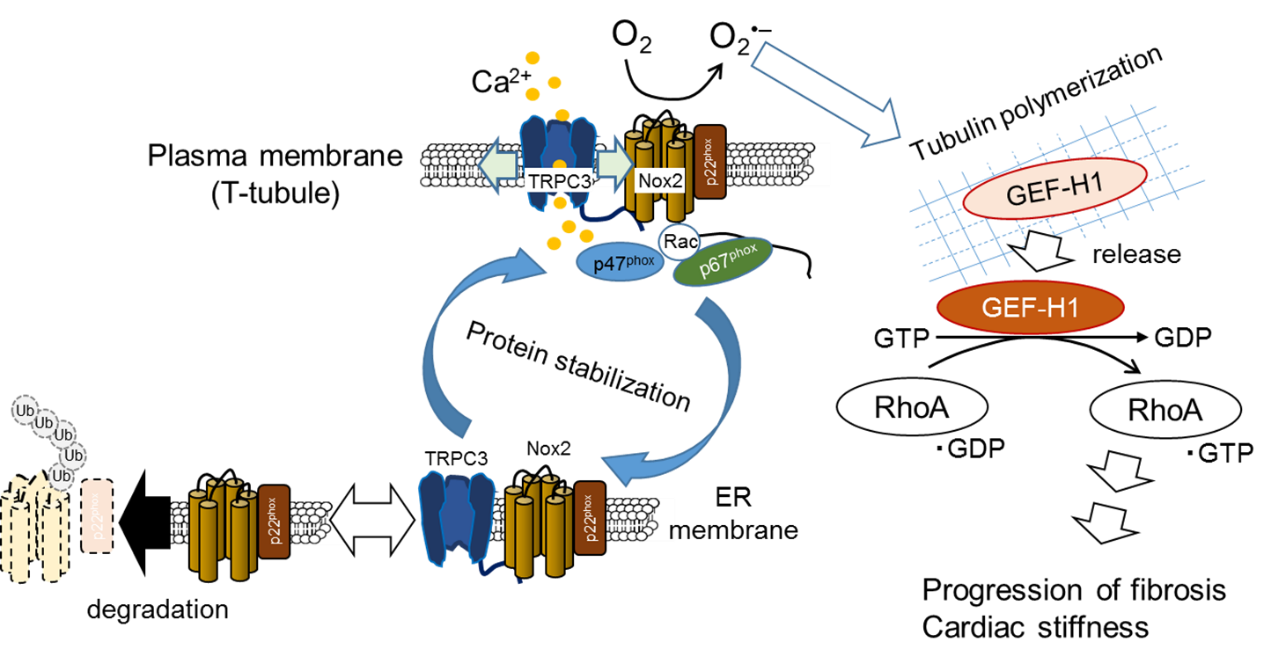
Heart failure is one of the major leading causes of morbidity and mortality in worldwide. Oxidative stress caused by excess accumulation of reactive oxygen species (ROS) have been suggested to mediate the development of structural and morphological changes of the heart (cardiac remodeling) induced by several risk factors including diabetic mellitus, hypertension and myocardial infarction. In the heart, there are two major ROS-producing pathways: the mitochondrial electron transport chain and the enzymatic functions of NADPH oxidase (Nox). Mitochondria are definitely the major source of ROS production involved in the pathogenesis of heart failure, but several studies have shown that inhibition of the Nox2 enzyme or Nox2 activators, such as Rac1 and p47phox, suppresses oxidative stress and cardiac dysfunction in mice with heart failure.
Transient receptor potential (TRP) family proteins, first described in a Drosophila visual transduction mutation trp, comprise 28 mammalian cation channels expressed in almost every tissue. Among them, canonical TRP subfamily (TRPC) proteins, two diacylglycerol (DAG)-activated TRPC members (TRPC3 and TRPC6), have been implicated in the development of pathological cardiac remodeling. TRPC3 and TRPC6 preferentially form hetero-tetramer channels and coordinately participate in angiotensin II-induced hypertrophic growth of neonatal rat cardiomyocytes (NRCMs) and pressure overload-induced cardiac hypertrophy in mice. We have recently reported using TRPC3-deficient mice that selective inhibition of TRPC3 is sufficient to attenuate pathological cardiac remodeling in mice. TRPC3 was found to positively regulate ROS signaling through increasing Nox2 protein stability by forming a protein complex with Nox2, supporting the pathological importance of TRPC3 in ROS-dependent heart failure. However, whether TRPC6 inhibition is sufficient to improve heart failure is still obscure.
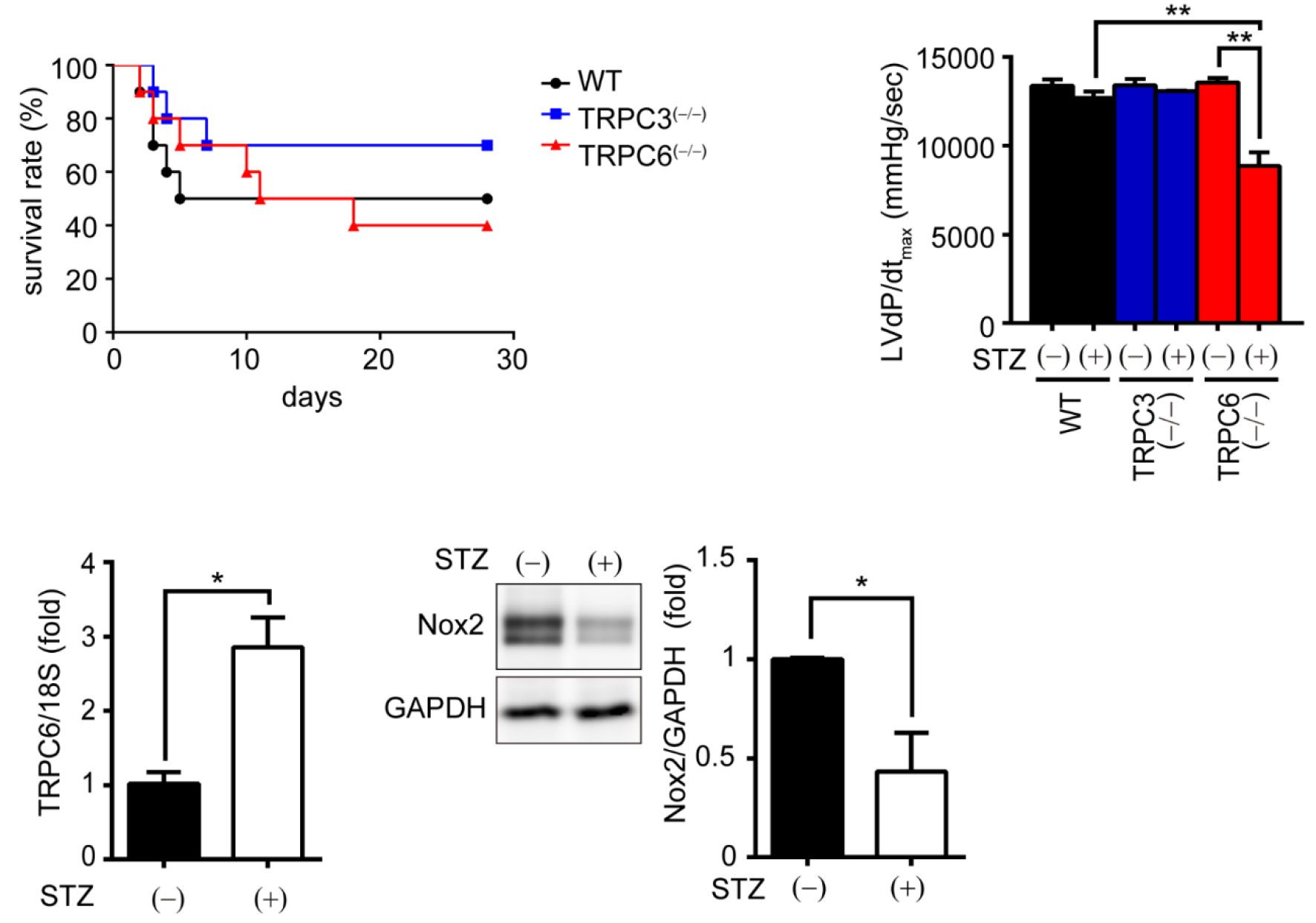
We here demonstrated that deletion of TRPC6 had no impact on pressure overload-induced heart failure despite inhibiting interstitial fibrosis in mice. TRPC6-deficient mouse hearts 1 week after transverse aortic constriction showed comparable increases in fibrotic gene expressions and ROS production but promoted inductions of inflammatory cytokines, compared to wild type hearts. Treatment of TRPC6-deficient mice with streptozotocin caused severe reduction of cardiac contractility with enhancing urinary and cardiac lipid peroxide levels, compared to wild type and TRPC3-deficient mice. Knockdown of TRPC6, but not TRPC3, enhanced basal expression levels of cytokines in rat cardiomyocytes. TRPC6 could interact with Nox2, but the abundance of TRPC6 was inversely correlated with that of Nox2. These results strongly suggest that Nox2 destabilization through disrupting TRPC3-Nox2 complex underlies attenuation of hyperglycemia-induced heart failure by TRPC6.
This research was funded by grants from the Japan Science and Technology Agency, Precursory Research for Embryonic Science and Technology Program (JPMJPR1336 to M.N.); Grants-in-Aid for Scientific Research (No. 25293018 and 16776376 to M.N.) and Scientific Research on Innovative Areas (Research in a Proposed Research Area ‘Oxygen Biology’ (No. 14430291 to M.N.) and ‘Living in Space’ (No. 16H01656 to T.N-T.) and Platform for Drug Discovery, Informatics, and Structural Life Science (to M.N.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Oda S., Numaga-Tomita T., Kitajima N., Toyama T., Harada E., Shimauchi T., Nishimura A., Ishikawa T., Kumagai Y., Birnbaumer L. and Nishida M., "TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice" Scientific Reports. (in press).
 [Ref.]
[Ref.]
Kitajima N. et al., TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 6, 37001; doi: 10.1038/srep37001 (2016).
Numaga-Tomita, T. et al., TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 6, 39383; doi: 10.1038/srep39383 (2016).
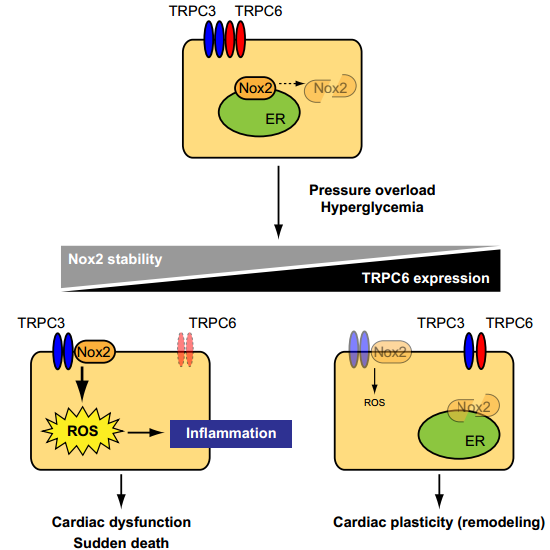
In resting condition, TRPC3 and TRPC6 channels function independently or coordinately in cardiomyocytes. Once hearts are exposed to environmental stresses such as hemodynamic load and hyperglycemia, TRPC3 forms stable protein complex with Nox2, which evokes aberrant ROS production in cardiomyocytes. In contrast, environmental stresses also upregulate TRPC6, which can counteract formation of the TRPC3-Nox2 complex in cardiomyocytes, leading to Nox2 destabilization, and resulting in negative regulation of ROS signaling in heart.
![]()
![]()
![]()
National Institute for Physiological Sciences
Kyushu University
Tsukuba University





