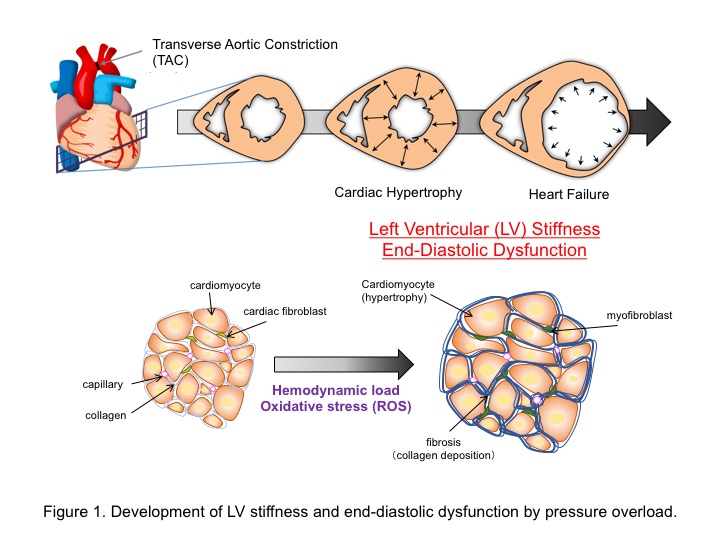
The heart comprises a highly dynamic mechanical environment that flexibly changes its structure and morphology to maintain its mechanical properties. Pressure overload-induced cardiac stiffness, defined by excessive accumulation of extracellular matrix components, is now attracting attention as a major cause of left ventricular diastolic dysfunction (Figure 1). However, how the heart decodes the mechanical stretch as a pathologic stress was obscure.
Reactive oxygen species (ROS) produced by NADPH oxidase 2 (Nox2), a microtubule-associated ROS-producing enzyme, function as key mediators of mechanotransduction during both physiological adaptation to mechanical load and maladaptive remodeling of the heart. This is despite low levels of cardiac Nox2 expression. Transient ROS production induced by mechanical stretch during diastolic filling triggers a burst of Ca2+ sparks through oxidative modification-dependent activation of ryanodine receptors. By contrast, persistent Nox2-derived ROS production in response to pressure overload in mice leads to oxidative stress through Nox2-derived ROS-induced ROS release from mitochondria and participates in the transition from cardiac adaptation to maladaptation. But how the heart alters mechanotransductive signaling against a background of rhythmic contraction and dilatation is obscure.We demonstrate that transient receptor potential canonical 3 (TRPC3), a Ca2+-permeable channel, acts as a positive regulator of ROS (PRROS) in cardiomyocytes, and specifically regulates pressure overload-induced cardiac stiffness, as well as interstitial fibrosis in mice. TRPC3 physically interacts with Nox2 at specific C-terminal sites, thereby protecting Nox2 from proteasome-dependent degradation and amplifying Ca2+-dependent Nox2 activation through TRPC3-mediated background Ca2+ entry. Nox2 also stabilizes TRPC3 proteins to enhance TRPC3 channel activity. Expression of TRPC3 C-terminal polypeptide abolished TRPC3-regulated ROS production by disrupting TRPC3-Nox2 interaction, without affecting TRPC3-mediated Ca2+ influx.
The novel TRPC3 function as a PRROS provides a mechanistic explanation for how diastolic Ca2+ influx specifically encodes signals to induce ROS-mediated maladaptive remodeling and offers new therapeutic possibilities.
This research was funded by grants from the Japan Science and Technology Agency, Precursory Research for Embryonic Science and Technology Program (to M.N.); Grants-in-Aid for Scientific Research (No. 25293018 and 16H05092 to M.N.; No. 12J05497 to N.K., No. 26670144 to T. N-T.) and Scientific Research on Innovative Areas (Research in a Proposed Research Area ‘Oxygen Biology’ to M.N. and ‘Living in Space’ to T.N-T.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; Daiko Foundation and Naito Memorial Foundation (to M.N.).
Naoyuki Kitajima, Takuro Numaga-Tomita, Masahiko Watanabe, Takuya Kuroda, Akiyuki Nishimura, Kei Miyano, Satoshi Yasuda, Koichiro Kuwahara, Yoji Sato, Tomomi Ide, Lutz Birnbaumer, Hideki Sumimoto, Yasuo Mori and Motohiro Nishida. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 6, 37001; doi: 10.1038/srep37001 (2016).
Numaga-Tomita, T. Naoyuki Kitajima, Masahiko Watanabe, Takuya Kuroda, Akiyuki Nishimura, Kei Miyano, Satoshi Yasuda, Koichiro Kuwahara, Yoji Sato, Tomomi Ide, Lutz Birnbaumer, Hideki Sumimoto, Yasuo Mori, Motohiro Nishida
TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 6, 39383; doi: 10.1038/srep39383 (2016).





